The FDA Regulatory Readiness and Drug/Device Development Tool (DDT/MDDT)
Update: TED and TRACK-TBI Investigators Respond to FDA's Request for Information on Identifying Potential Biomarkers
In a collaborative effort, TED and TRACK-TBI Investigators drafted two responses to the FDA's Request for Information on Identifying Potential Biomarkers for Qualification and Describing Contexts of Use to Address Areas Important to Drug Development. Responses on the potential of blood-based and MRI biomarkers in TBI were submitted.
To read the response on Blood-Based Biomarkers, click here.
To read the response on MRI Biomarkers, click here
Qualification Pathways
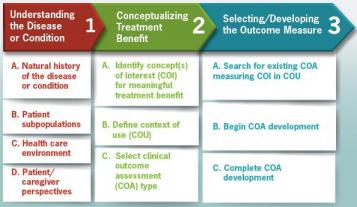
Fig. 1. FDA roadmap to patient-focused measurement in clinical trials
FDA Guidance

Fig. 2. Categories of Biomarkers for DDTs
DDTs include, but are not limited to, clinical outcome assessments (COAs) and biomarkers. COAs, by the FDA’s definition, “…measure a patient’s symptoms, overall mental state, or the effects of a disease or condition on how the patient functions.” Biomarkers are defined by FDA as a “characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathologic processes, or biological responses to a therapeutic intervention.” FDA describes qualification as “a conclusion that within the stated COU, the DDT can be relied on to have a specific interpretation and application in drug development and regulatory review.”
On the FDA website, you can read more about the Guidance that manages the Clinical Outcome Assessment Qualification Program, another that manages the Biomarker Qualification Program, and a similar process is utilized for validation of medical devices.
Qualification has widespread implications for candidate therapies; it may confer broad applicability across multiple drug candidates, independent of the mechanism of action of the drug or of the contributing sponsor as well as across multiple clinical disorders, drugs, or drug classes. As well, qualified DDTs become open standards for the scientific community. Published recommendations describe the type and quality of data that must be submitted as part of CDER’s formal DDT qualification process.
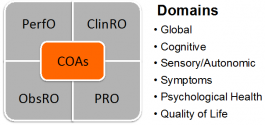
Fig. 3. COAs and Domains for Diagnostic and Therapeutic Trials
Center for Drug Evaluation and Research Organization (CDER) Critical Path Innovation Meeting (CPIM)
The Critical Path Innovation Meeting (CPIM) is an additional portal to FDA collaboration on the regulatory pathway. CPIM was introduced by CDER in fall 2014 “to address issues in drug development identified in the 2004 FDA publication, Innovation or Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products Challenges and Opportunities Report.” As articulated by CDER:
The CPIM mechanism encourages communication and engagement between CDER and investigators across the private-public spectrum to improve efficiency and success in drug development. Among the topics a CPIM might include are discussion of a methodology or technology proposed by the meeting requester and CDER’s general advice on how this methodology or technology might enhance drug development.
FDA Letter of Support
Another area of engagement in the pre-qualification stage of the drug development process is CDER’s Letter of Support, which may be issued following submission of preliminary evidence by a drug development tool proponent that briefly describes CDER’s observations as to the potential value of a biomarker and encourages further evaluation. As CDER notes, “This letter does not connote qualification of a biomarker. It is meant to enhance the visibility of the biomarker, encourage data sharing, and stimulate additional studies.”